La nitración es una clase general de proceso químico para la introducción de un grupo nitro en un compuesto orgánico. Se trata de la reacción entre un compuesto orgánico y un agente nitrante (por ejemplo el ácido nítrico) que introduce un grupo nitro en el hidrocarburo produciendo un éster. La nitración es una de las reacciones químicas comercialmente más importantes.
Grupo Nitro
NO2+ (ión Nitronio)
Historia
Los compuestos nitro aromáticos más tempranos se obtuvieron por Mitscherlich en 1834 por el tratamiento de hidrocarburos derivados del alquitrán de hulla con ácido nítrico fumante. Por 1835 Laurent estaba trabajando en la nitración de naftaleno, el hidrocarburo aromático más fácilmente disponible puro en ese momento. Dale informó sobre compuestos nitro mixtos derivados de benceno crudo en la reunión anual de la Asociación Británica para el Avance de la Ciencia de 1838. Sin embarg no fue sino hasta 1845 que Hofmann y Muspratt informaran de su trabajo sistemático en la nitración de benceno para dar mono y dinitrobencenos mediante el uso de una mezcla de ácidos nítrico y sulfúrico.
La primera producción en pequeña escala de nitrobenceno se destiló cuidadosamente para dar un líquido amarillo con un olor a almendras amargas que se vendía a fabricantes de jabón y perfume como "esencia de mirbano."
El proceso de reducción de hierro de Bechamp, que hizo la anilina más fácilmente disponible, se publicó en 1854, y el descubrimiento de la anilina malva por Perkinin 1856 comenzó la industria europea de colorante de anilina que se convirtió en la base para una industria de colorantes sintéticos que en todo el mundo se estima que han tenido ventas de $ 6 × 109 en 1988.
El desarrollo de procesos y aumento a escala de los procesos de nitración y reducción, iniciada por Perkin fue continuada por muchos otros. En 1985 la producción europea de anilina fue de 500 000 t / a, y sigue siendo el mayor producto basado en un proceso de nitración. Su uso en colorantes ahora representa sólo el 4% de la producción de anilina, porque la mayor parte del crecimiento se ha debido a las gomas químicas y los isocianatos, este último ahora consume más del 50% de la producción de nitrobenceno para la fabricación de difenildiisocianato metileno.
Tipos de nitración
De acuerdo a la estructura química del producto nitrado, la nitración puede clasificarse como:
1) C - Nitración
Donde un grupo nitro ( NO2+ ) es ganado por un átomo de carbono como se muestra

2) O - Nitración
Resultando la formación de un nitrato
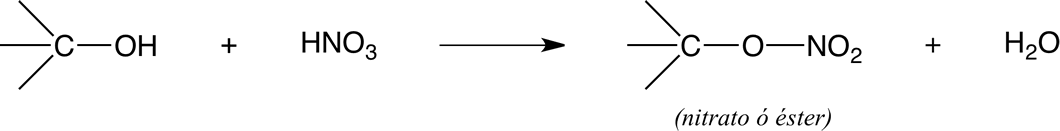
Reacción típica conocida como esterificación.
3) N - Nitración
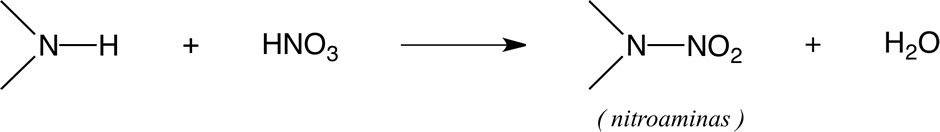
Es el producto de la sustitución de un átomo de hidrógeno por un grupo nitro produciendo nitroaminas como en el ejemplo.
En cada uno de los ejemplos indicados, un grupo nitro reemplaza a un átomo de hidrógeno. Sin embargo el grupo nitro puede reemplazar a otros átomos o grupos de átomos, la reacción de Víctor Meyer es un ejemplo típico, donde un átomo halógeno (especialmente bromo o iodo) es reemplazado por un grupo nitro usando nitrito de plata (o nitrito de sodio).
La O-nitración para dar nitratos y N-nitración para dar nitraminas son de mucho menor importancia comercial para los compuestos aromáticos que la C-Nitración.
Los compuestos nitrados pueden ser producidos por determinadas reacciones de adición como ser ácido nítrico, o bióxido de nitrógeno con compuestos orgánicos no saturados. Por ejemplo Olefinas o Acetilenos.
El proceso de nitración es altamente exotérmico, entregando al medio más de 30 Kcal / mol. El calor de reacción, no obstante, varía con el hidrocarburo a nitrar. La reacción de nitración es siempre fuertemente exotérmica, como se ejemplifica en la mononitración de benceno (H = - 117 kJ / mol) y naftaleno (H = - 209 kJ / mol), y es probablemente la unidad de proceso más potencialmente peligrosa operada industrialmente. Esto se debe fundamentalmente a que el calor generado, lo que puede desencadenar el poder de ácido nítrico para degradar materiales orgánicos exotérmicamente a productos gaseosos con violencia explosiva. Compuestos nitroarilo, especialmente aquellos con más de un grupo nitro, son potencialmente peligrosos debido a sus muy altos contenidos de oxígeno. Algunos compuestos polinitro (por ejemplo, trinitrotolueno y ácido pícrico) son detonables y tienen una larga historia de uso como explosivos. Un peligro menos publicitado con compuestos nitroaromáticos es ocasionada por las violentas reacciones de descomposición que pueden ocurrir al calentar con un álcali.
El mecanismo de nitración depende de los reactivos y las condiciones de operación.
Las reacciones pueden ser del tipo iónico ó del tipo radicales libres.
El tipo iónico se usan comúnmente en la nitración de hidrocarburos aromáticos, alcoholes simples glicoles, gliceroles, celulosas y aminas.
El tipo radical libre se da en nitración de parafinas, cicloparafinas y olefinas, los compuestos aromáticos y algunos hidrocarburos pueden algunas veces ser nitrados reaccionando con radicales libres, pero generalmente con menos éxito.
Para estas reacciones de nitración frecuentemente son usados catalizadores sólidos.
Los productos principales obtenidos por nitración son: derivados de celulosa, explosivos, nitrobenceno, nitrotolueno, nitrofuranos, nitroparafinas, nitrofenoles, nitrocelulosa, etc.
Nitración Tipo Iónico
Muchas nitraciones iónicas emplean ácidos mezcla, la que usualmente contiene ácido nítrico + un ácido fuerte, por ejemplo ácido sulfúrico / ácido perclórico / ácido hidrofluórico / resinas de intercambio iónico conteniendo grupos de ácido sulfónico utilizan al agente nitrante en solución líquida.
En muchos casos se presentan 2 fases inmiscibles (líquidas) dentro del reactor, se mantiene una leve presión sobre la atmosférica para conservar las dos fases líquidas. En la fase orgánica se concentran los nitrohidrocarburos. En la fase ácida se concentra el agua. El lugar de la nitración propiamente dicha está junto a la interfase.
Por razones de seguridad la operación es :
- A presión atmosférica
- Rango de temperaturas [0 a 120] ºC por lo tanto el rendimiento será función de la temperatura = f (T)
Este tipo de nitración aumenta el tiempo de residencia (oscila alrededor de 1 hora). Además no se deben utilizar temperaturas menores a 0 ºC dado que el rendimiento bajará y se incrementará el costo de refrigeración.
Agentes de nitración iónica
Frecuentemente se utiliza HNO3 (HO.NO2)
Por razones de viabilidad y economía la nitración a escala industrial se lleva a cabo habitualmente con una mezcla de ácidos nítrico y sulfúrico (mezcla sulfonítrica), y de vez en cuando con ácido nítrico acuoso, ácido nítrico en ácido acético, o ácido nítrico en anhídrido acético. El uso de ácidos de componentes alternativos, tales como ácido perclórico, ácido fluorhídrico, o trifluoruro de boro se limita a importantes estudios de soporte. Estos a veces son llevadas a cabo en disolventes orgánicos inertes tales como hidrocarburos clorados o sulfolano para dar mezclas de reacción homogéneas.
La verdadera especie nitrante en reacciones tipo iónica es el nitronio NO2+
El ácido nítrico puro, ionizado produce pequeñas cantidades de ión nitronio.
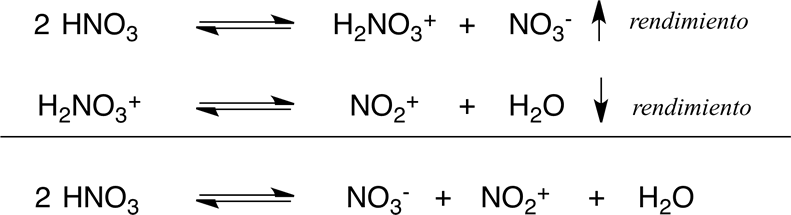
La primera reacción es rápida y la segunda lenta.
La presencia de H2O (producida durante la nitración) disminuye la concentración del ión [NO2+] (=> desplazamiento de la reacción hacia la izquierda).
La eficiencia de nitración es mayor usando ácido sulfúrico H2SO4 ú Oleum (SO3 + H2SO4)
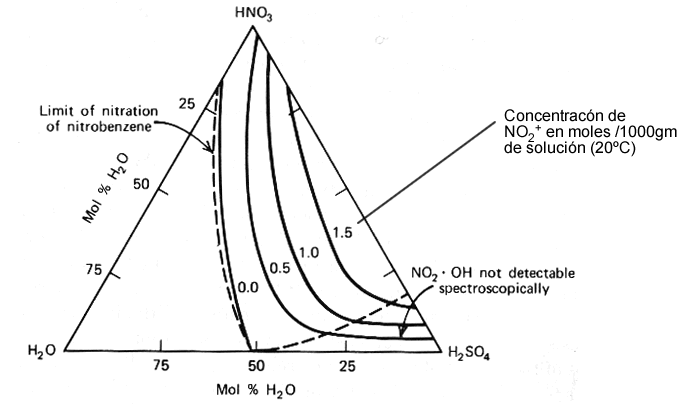
Figura 1
A altas concentraciones de ácido sulfúrico, la mayor parte de ácido nítrico se ioniza
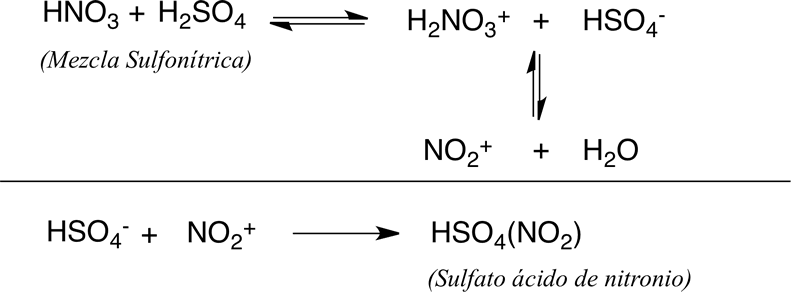
El ión de nitronio, NO2 + , se considera que es la especie activa en todos estos sistemas. En el sistema más común la ecuación general
HNO3 + 2H2SO4 ⇌ NO2+ + H3O+ + 2HSO4-
es un compuesto de muchos equilibrios presentes en las mezclas de HNO3 - H2 SO4 - H2O. Todos ellos deben tenerse en cuenta al considerar la reactividad del sustrato y el grado de nitración que se requiere.
La figura 1 indica que el aumento de agua disminuye la concentración del ión nitronio hasta concentraciones molares del 50 %.
Concentraciones mayores del 50 % no detectan ión nitronio.
Concentraciones mayores de ácido nítrico, en algunos casos incrementan la concentración del ion nitronio, pero a la vez la fracción de ácido nítrico que es ionizado decrece.
Para ácido nítrico puro aprox. sólo el 3% en peso de éste es ionizado a la forma del ión nitronio.
El ácido sulfúrico es por lejos el ácido fuerte más comúnmente usado en mezcla de ácidos, además de barato y altamente efectivo en promover la formación de NO2+.
Pero a la vez tiene la desventaja que la formación de agua lo diluye y no se conoce un método simple de separación completa del agua del ácido sulfúrico.
La separación parcial del agua del ácido sulfúrico se efectúa removiendo el desperdicio con vapores. El ácido remanente es concentrado aprox. al 93% con combustión de gases y finalmente se obtiene 98% por adición de trióxido de azufre.
El ácido suficientemente concentrado, apto para la reacción de nitración, se recicla y el ácido adicional equivale a la cantidad de trióxido de azufre adicionado.
El rol del ácido sulfúrico fue previamente considerado como un agente deshidratante capaz de remover el agua producida durante la nitración, mientras ésta iba completándose en forma fría.
Un agente de nitración aromática típico para mononitración a gran escala consiste en 20% de ácido nítrico, 60% de ácido sulfúrico, y 20% de agua; esto se conoce como mezcla sulfonítrica 20/60/20. Una definición alternativa del mismo ácido, utilizado en algunas situaciones, es ácido nítrico 15% molar, ácido sulfúrico 30% molar y agua 55% molar. Comúnmente, juntos el sustrato aromático líquido y el producto nitrado forman una fase separada de la de ácido mixto acuosa. Por lo tanto, se requiere una agitación eficiente para maximizar el contacto con la fase orgánica y minimizar la resistencia a la transferencia de masa. Sustratos sólidos se disuelven mejor en la fase de ácido sulfúrico. Ácido nítrico fuerte conduce a reacciones secundarias oxidativas, mientras que una temperatura más alta conduce a la disminución de la concentración de iones de nitronio. Se requiere mucho trabajo de desarrollo detallado en cada nitración individual para optimizar estas y otras variables, para maximizar la formación del isómero requerido, y para minimizar las reacciones secundarias.
Cinética de Nitración de Aromáticos
La cinética puede variar en forma considerable, desde segundos a varias horas. La composición del ácido, la temperatura y el grado de agitación son los principales factores. Una agitación vigorosa eliminaría las resistencias a la transferencia de masas en los sistemas de 2 fases.
Las constantes de cinética aumentan con la temperatura, las solubilidades en la fase ácida de los compuestos nitrados ó aún no nitrados, probablemente, también aumenten con la temperatura. A altas concentraciones de H2SO4 , la solubilidad del ácido nítrico en la fase orgánica es baja. Además la viscosidad disminuye con la temperatura, ósea que los coeficientes de difusividad aumentan. El tamaño de la gota impulsada por el agitador, también incide en la cinética. Gotas de tamaño muy pequeño, impulsados a altas velocidades disminuyen la transferencia de masa del reactivo hacia la fase dispersa. La composición del ácido y las fases orgánicas afectan también la tensión superficial entre las fases. Esta relación (ácido/fase orgánica) es importante por el tipo de emulsión que se forma y por el área entre fases.
Si la temperatura sube demasiado, el grado de exotermia de la reacción puede volverse fuera de control, produciéndose reacciones laterales ó explosiones.
Se emplean reactores que están compartimentados en serie con un eficiente agitador en cada uno, tal el proceso para el TNT ó la Nitroglicerina.
Otros poseen un tubo principal y 2 laterales; la emulsión es reciclada muchas veces por el primero con grandes zonas de transferencia de calor.
La recirculación ocurre mediante una bomba en el fondo de cada tubo lateral, que a la vez provee la agitación y mezcla de los reactivos.
Desventajas
- Ácido sulfúrico diluido
- Concentración ácido para venta ó reuso ⇒ aumento del costo energía calórica
- Costo de tratamiento para eliminar vestigios de materia orgánica.
- Idem anterior p/restos nítrico y nitroso.
- Nunca se consigue mejor calidad por altísimos costos. (Rango [ ] de 90 a 98 %)
- Altos costos de instalaciones de tratamiento
Ventajas
- Rol deshidratante del S O3 (remueve agua de reacción)
- Promotor de la ionización HNO3 → NO2+ ( se comporta como catalizador)
- Ventas del subproducto (auge minería, lixiviación)
Algunas nitraciones como C-Nitración son altamente irreversibles. La energía libre de Gibbs en nitración es grande y negativa.
El ácido sulfúrico actúa primariamente como catalizador provocando la ionización del ácido nítrico.
La composición de equilibrio para reacciones entre ácido sulfúrico y ácido nítrico, tanto como las reacciones O-Nitración y N-Nitración son presumiblemente función de la temperatura.
Algunas veces se emplean mezclas de bióxido de nitrógeno o nitratos alcalinos con ácido sulfúrico( u otro ácido fuerte).
La ionización puede ocurrir y NO2+ y / o NO+ están presentes en muchas mezclas.
El ácido acético y el anhídrido acético son algunas de las combinaciones que con ácido nítrico forman mezclas que son altamente efectivas para facilitar el proceso de nitración.
Esto hace surgir una pregunta: ¿Cómo el ácido acético, un ácido relativamente débil, forma NO2+ en forma análoga al ácido sulfúrico?
Esto se explica con el siguiente mecanismo:
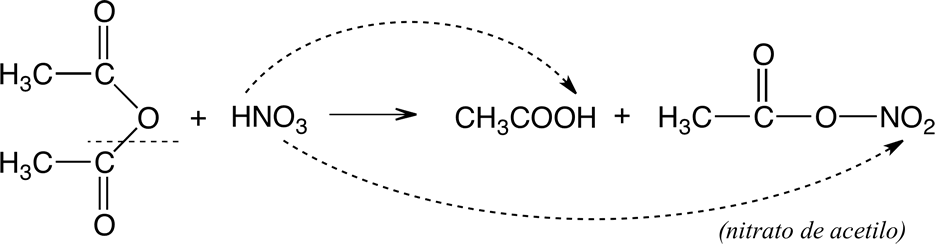
Es muy utilizado en la nitración comercial de hexametilentetraamina (CH2 ) 6 N4 para obtener ciclotrimetilentrinitroamina.
Un rol adicional para el anhídrido acético ó ácido acético es que tiende a incrementar la solubilidad de las 2 fases.
Mezclas de nítrico y nitroso se usan para nitraciones suaves de aromáticos (en determinadas condiciones de los reactivos fenol o anisol).
El ión NO+ (nitrosilo) es considerado iniciador pero de baja reactividad. Por excelencia se usa NO2+ .
Reacciones de Nitración tipo iónico
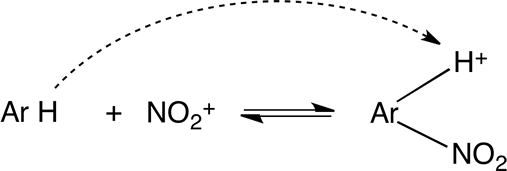
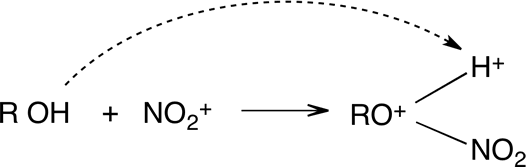
Los compuestos aromáticos, alcoholes y aminas reaccionan con NO2+ formando complejos relativamente inestables , como se muestra :
Aromáticos
Alcoholes
Aminas
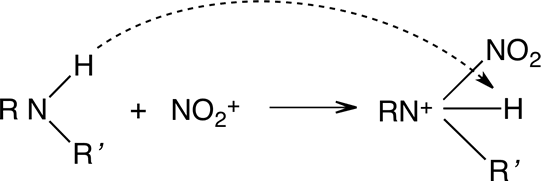
Estos complejos se forman lentamente pero se descomponen rápidamente en medio ácido (aniónico, HSO4- ,NO3- ).
El protón H+ abandona el complejo formando el producto nitrado y regenerando el "catalizador" con el anión (HSO4- + H+ ⇌ H2SO4).
Nitración de Compuestos Aromáticos
Si poseen grupos alquilos (tolueno, etilbenceno) son más reactivos que el benceno.
El grupo alquilo incrementa la densidad electrónica del anillo en posiciones orto y para.
Como resultado el NO2+ tiende a nitrar estas posiciones del anillo.
Los aromáticos que reaccionan con grupos nitro forman mononitrobenceno o mononitrotolueno y son más difíciles de nitrar que los hidrocarburos no aromáticos.
Si el grupo es por ejemplo: —CF3- , los electrones se concentran en las posiciones meta y para.
Una vez hecha la 1o nitración (mono), resulta más dificil introducir el 2º grupo nitro (dinitración) y así progresivamente para la 3ª.
Condiciones muy severas, mayor temperatura y mayor riesgo. También requieren mayor [ácidos] ⇒ mayor [NO2+].
Se producen reacciones secundarias y subproductos di-trinitrados que se convierten en impurezas a eliminarlas por razones de seguridad en los siguientes pasos.
Aromáticos como fenoles, éteres fenólicos, fáciles de nitrar, son nitrados con mezclas de ácidos nítricos y un plus mayor de ácido nitroso. La ionización de esta mezcla produce el ión nitrosilo (NO+) como sigue:
HNO3 + HNO2 ⇌ NO+ + NO3+ + H2O
El ión nitrosilo ataca al compuesto aromático
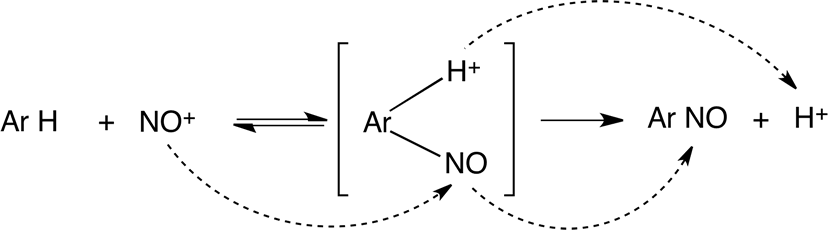
El aromático nitroso es oxidado con ácido nítrico formando nitroaromáticos y volviendo a formar ácido nitroso.
Ar NO + HNO3 → Ar NO2 + HNO2 (regeneración)
2- Nitración de Alcoholes
• O - Nitración
• Utiliza mezcla ácidos como agente nitrante. Estas reacciones son complicadas porque
son reversibles y frecuentemente incluyen 2 fases.
• En la mayoría de los casos utilizan temperaturas bajas.
Ejemplo : GLICEROL
Es nitrado a 20 / 35oC produciendo glicerina trinitrada (trinitro glicerina ).
CH2 OH CH2 — O — NO2
| |
CH OH + 3 NO2+ → CH — O — NO2
| |
CH2 OH CH2 — O — NO2
triNitroglicerina
Se evitan altas temperaturas → riesgo de explosión y razones de equilibrio térmico.
Glicoles y alcoholes en general son nitrados en condiciones similares al glicerol.
También la celulosa (C6 H10 O5) es nitrada por O--Nitración obteniendo nitrato de celulosa, usando como agente nitrante mezcla de ácidos nítrico y sulfúrico.
En muchas nitraciones comerciales están presentes las 2 fases líquidas, una fase ácida o acuosa y la otra orgánica. Muchas reacciones de nitración se ubican en las fase ácida y solo el 10% ocurre en la fase orgánica. Sin embargo las reacciones de oxidación ocurren en la fase orgánica en mayor grado que en la ácida.
Nitraciones Tipo Radical Libre
Sedivide en:
- Nitraciones en fase vapor
- Nitraciones en fase líquida.
Nitración En Fase Vapor
- Baja presión (para obtener mezclas gaseosas de hidrocarburos, agentes nitrantes, productos e inertes)
- Cuando se usa HNO3 como agente nitrante el rango de temperaturas es de 350 a 450 ºC.
- Cuando se usa dióxido de Nitrógeno → temperatura ∈ ( 200 a 450 ) ºC
- En altas temperaturas la reacción es instantánea; a bajas temperaturas aumenta el tiempo de reacción para obtener un adecuado grado de nitración llegando al orden de varios minutos.
- Mayor exotermia, mayor temperatura ⇒ ↑↑ riesgo de explosividad ⇒ estricto control de temperaturas.
Los procedimientos comunes para el control de las temperaturas son :
- Usar exceso de hidrocarburos, 4:1 con relación del hidrocarburo al agente nitrante; el cual actúa disminuyendo la temperatura.
- Adición de un gas inerte (vapor, N2 ) a la mezcla que reacciona. A veces se usa ácido nítrico diluido (60%) formándose grandes cantidades de vapor.
- Diseño de una adecuada transferencia de calor, tubos de aluminio de diámetro pequeño en baño de sal fundida.
- Reactores de lecho fluido también proveen un excelente control de temperatura.
- Empleo del calor de reacción por vaporización, calefaccionando la alimentación de ácido nítrico usado como agente nitrante. este es pulverizado dentro de un vapor propano caliente.
Las presiones varían entre [1 , 12] atm.
En nitraciones en fase vapor de parafinas usando ácido nítrico, resulta máxima la conversión de éste a varias nitroparafinas, desde cerca del 20 % en el caso del metano a aprox. el 40 % para parafinas altas como el n-butano. El ácido nítrico residual se convierte en óxidos de nitrógeno
Las conversiones crecen conforme aumenta el peso molecular de las parafinas.
El metano y el etano que contienen 1 ó 2 átomos de carbono primario, son más difíciles de nitrar que el propano y parafinas con átomos de carbono secundario.
La conversión de dióxido de nitrógeno a nitroparafinas son significativamente menores que las basadas en ácido nítrico.
Teóricamente las máximas conversiones son 67% como las indicadas en la siguiente reacción estequeométrica:
2RH + 3NO2 → 2RNO2 + H2O + NO
100% 67% 33%
Con NO2 ⇒ conversiones moderadas
Ejemplo: máxima conversión en nitración con NO2 al propano (C3 H8 ) es de 27 %.
El O2 ↑ η (1ó2 molesdeO2 /moldeHNO3 ),elincrementopuedellegar a un 50 % de > η .
También pueden usarse halógenos (Br2 , Cl2 ) según 0.003 y 0.06 moles respectivamente por cada mol de ácido nítrico pero ocasionan problemas de corrosión.
Se produce una considerable cantidad de subproductos: aldehídos, CO, CO2 , H2O, parafinas livianas, alcoholes y cetonas.
Cuando no usamos aditivos menos de la mitad del ácido nítrico es convertido a nitroparafinas, el resto forma óxido nítrico además de cantidades menores de dióxido de nitrógeno, óxido nitroso y nitrógeno.
En la industria el óxido nítrico y el dióxido de nitrógeno son recuperados y reformados a ácido nítrico. El resultado neto es que del 60 % al 80 % del ácido nítrico es convertido a nitroparafinas.
Nitración En Fase Liquida
La nitración en fase líquida de parafinas ocurre predominantemente por medio de radicales libres en la reacción.
• Mezcla de ácidos altamente ionizados, no son efectivos reaccionando con hidrocarburos no polares.
• Si es efectivo el HNO3 no mezclado (puro).
• Elrangodetemp.ε (100,200)ºC
Suficiente presión para mantener los reactivos inmersos en la fase líquida (4 a 200 atm). Se necesitan altas presiones para mantener el óxido nítrico y el nitrógeno en fase gas disueltos en los líquidos. La mínima presión estará dada por la volatilidad de la parafina nitrándose. Presiones muy superiores, no tendrán efecto en la reacción.
• Al mantenerse la presión correspondiente → 2 fases (líquido + vapor ) → aumenta la corrosión del reactor.
Las fracciones molares de hidrocarburos varían desde 2 : 1 a 6 : 1 respecto de los agentes nitrantes. Se usa el ácido nítrico concentrado al 60 / 70 % como agente nitrante , tomando entonces importancia el grado de agitación y el carácter de la emulsión.
También se usa como agente nitrante el NO2 que es relativamente soluble en hidrocarburos, formando una fase líquida hasta la fase acuosa. Las mayores reacciones probablemente ocurran en la fase orgánica, dado que el NO2 es la principal especie nitrante aún si se use ácido nítrico.
A las temperaturas usadas en fase líquida el tiempo de residencia en reactores tubulares oscila de 1 a 4 minutos con el objeto de obtener de un 10% a un 20% más de transformación de agentes nitrantes a nitroparafinas.
El dióxido de nitrógeno es pensado como el verdadero agente nitrante y en los casos más frecuentes el ácido nítrico suele descomponerse en dióxido de nitrógeno.
Mecanismo de las reacciones de nitración
A. Reacciones del ácido nítrico
A1. HNO3 → NO2+ + OH+
A2. HNO3 + NO → HNO2 + NO2+
ó también
HNO3 + NO →NO + OH- + NO2+
B. Reacciones de parafinas (RH)
B1. RH + OH-- → R-- + H2O
B2. RH + NO2 → R-- + HNO2 → R- + OH- + NO
B3. RH + O2 → R-- + ONO-
El oxígeno puede obtenerse por la descomposición del ácido nítrico o dióxido de nitrógeno.
Reacción que produce radicales alquilo usando ácido nítrico : La ecuación que se obtiene sumando A1 + 2 x B1 + B2 =>
3RH + HNO3 → 3R-- + H2O + NO
Reacción que produce radicales alquilo usando dióxido de nitrógeno :
2RH + NO2 → 2R-- + H2O + NO
Obtenida por la suma de B1 y B2
C. Reacciones de Radical Alquilo
1. R-- + R ONO H NO3 → + OH- (indudablemente un paso importante en la fase vapor de nitración con ácido nítrico)
2. R-- +HNO3 →RO-+HNO2→RO-+OH-+NO
3. R-- + NO2+ → RNO2 Paso importante en la nitración con dióxido de nitrógeno
4. R-- + ONO-- → R ONO (nitrito inestable)
5. R-- + OH - → R OH ( probablemente de menor reacción)
6. R-- + OH- → Olefina + H2O (puede haber otras reacciones sustrayendo un átomo de hidrógeno de un radical alquilo)
7. R-- + R' → RR' (así se pueden formar alcanos superiores)
8. R-- + O2 → R OO-- (radicales alquilo peróxido reaccionan formando compuestos oxigenados)
D. Descomposición de nitritos alquilo
1. RONO → RO- + NO
Los nitritos alquilo son muy inestables especialmente a altas temperaturas.
Esta reacción es fundamental para la producción de radicales Alqoxi a 350/450 oC (temperaturas cuando se usa ácido nítrico como agente nitrante ).
Cuando se usa dióxido de nitrógeno a 200 / 300 oC algunos nitritos alquilo pueden ser descompuestos .
Se obtienen cantidades significativas de ciclohexilnitrito nitrando ciclohexano con dióxido de nitrógeno a 200/240 ºC.
E. Reacciones de radicales Alqoxi
Es la producción de compuestos oxigenados como aldehídos, alcoholes, cetonas, etc.
La unión C C frecuentemente se rompe formando radicales alquilo más cortos. Se usan radicales propilo como los siguientes:
1. CH3 CH3
CH2 → CH2 + CH2O
CH2O-- radical etilo formaldehído
2. CH3
HCO-- → CH3-- + CH3
CH3 HCO--
radical metilo acetaldehído
3. CH3
HCO-- + OH-- → Acetona + H2O
CH3
El radical ciclohexiloxi reacciona formando ciclohexanona obteniendo cantidades significativas cuando se nitra el ciclohexeno. Posiblemente NO2 , ONO , etc., radicales también sustrae 1 átomo de hidrógeno.
4. RO-- + RH ( o aldehído u otro compuesto ) → R. OH ( alcohol) + R--
5. RO-- + NO2 → RONO2 (nitrato)
El ciclohexilnitrato se forma en cantidades significativas durante la nitración del ciclohexano a 200 / 240 ºC
6. RO-- + O2 → Productos Oxigenados
Los aldehídos , cetonas , alcoholes , etc. continúan oxidando a ácidos , óxidos de carbono , agua , etc.)
Química De La Reacción Tipo Radical Libre.
La química de los radicales libres es complicada por las reacciones de nitración y oxidación que se cruzan. En nitración con ácido nítrico, un mecanismo en cadena ocurre frecuentemente, iniciando, propagando y terminando la reacción . Todos los pasos son importantes dependiendo de la nitración específica.
Cuando se usa ácido nítrico en fase vapor, la reacción A.1 representa el paso principal (descomposición del ácido nítrico). Para iniciar este paso se requieren temperaturas del orden de 350oC y esta representa la velocidad de control de los pasos siguientes. Las reacciones de formación de radicales alquilo C.1 y nitroparafinas B.1 son pasos importantes en la cadena de nitración.
Cuando se usa dióxido de nitrógeno, las reacciones principales son B.2 (paso inicial a la formación de un radical libre) y C.3 (producción de nitroparafinas) éstas reacciones ocurren fácilmente a temperaturas cercanas a los 200 ºC.
La temperatura exacta depende del hidrocarburo específico a ser nitrado y presumiblemente B.2 sea la reacción control en este caso. La reacción C.3 es de menor importancia en nitraciones con ácido nítrico. La cinética de A.1es la más lenta de todas las reacciones de nitración.
Una reacción importante en todo radical alquilo que es nitrado es la C.4 en la cual se forma un compuesto inestable. Este nitrito parcial o totalmente descompuesto forma óxido nítrico y un radical alqoxi, en las reacciones E.1 a E.6 forma compuestos oxigenados y radicales alquilo de bajo peso molecular. Estos radicales pueden formar nitroparafinas de bajo peso molecular por medio de las reacciones C.1 a C.3. Los hidrocarburos oxigenados frecuentemente vuelven a reaccionar produciendo incluso ligeros productos oxigenados, óxidos de carbono y agua. Adicionando tanto halógenos como oxígeno, los anteriores reaccionan produciendo radicales alquilo. El incremento de concentración de estos radicales permiten conversiones más altas a nitroparafinas utilizando como agente ácido nítrico. Las reacciones B.2 y C.3 son pasos importantes para la fase líquida de nitración de parafinas. El óxido nítrico producido es oxidado con ácido nítrico para reconvertirse a dióxido de nitrógeno, el cual continúa la reacción. El proceso es complicado por el hecho que 2 fases líquidas están siempre presente y como resultado el óxido de nitrógeno tiende a transferirse de una fase a otra. La reacción C.9 (de un grupo alquilo con el óxido nítrico formado durante la reacción) es importante en la nitración en fase líquida. El compuesto nitroso formado puede volver a formar un oxido el cual reacciona con el dióxido de nitrógeno dando dinitroparafinas en la cual ambos grupo nitro son anexados a átomos de carbono. Dinitroparafinas como 2-2 dinitropropano puede ser producido fácilmente por un proceso en fase líquida pero no a altas temperaturas en nitración en fase vapor. Si se formaran a altas temperaturas se descompondrían rápidamente. Esto también es posible a través de la reacción C.9 con baja probabilidad a alta temperatura o en fase vapor cuando las concentraciones de los componentes son bajas comparada con el proceso en fase líquida.
Consideraciones De Seguridad
Numerosos peligros están presentes en muchas reacciones de nitración y en el manipuleo de productos nitrados . Se sabe que muchos hidrocarburos nitrados son explosivos, por ejemplo el trinitrotolueno, el ácido pícrico, la nitroglicerina (glicerol trinitrado) y algunas celulosas nitradas Dinitrotolueno y dinitro benceno son también explosivos.
La mezcla de nitrobenceno, ácido nítrico y agua son explosivos bajo un extenso rango de condiciones.
El nitrometano ha sido testado como monopropelente en cohetes y se despacha en pequeños tambores.
Los nitroaromáticos y algunas parafinas polinitradas son altamente tóxicas en el aire o en contacto con la piel, por ejemplo nitrobenceno y nitrotolueno.
Se deben tener en cuenta las precauciones de seguridad en el manipuleo de los agentes nitrantes y en la prevención de combustión en los más inflamables.
Nitrobenceno
El nitrobenceno es un líquido amarillo pálido el cual según la pureza puede variar a amarillo amarronando; con olor a almendras amargas, C6H5NO2,
Es soluble en la mayoría de los solventes orgánicos y miscible con dietil-eter y benceno en todas proporciones. Es parcialmente soluble en agua con 0,19 partes por 100 partes de agua a 20°C. Su densidad es de 1,2. El punto de ebullición a 101 kPa es de 210,9°C y a 0,13kPa 53,1°C.
Se elabora industrialmente por nitración directa del benceno usando la mezcla ácida de Nítrico + Sulfúrico. Dado que se forman 2 fases en la mezcla en reacción y los reactivos están distribuidos entre ellas, se controla el avance de la reacción mediante la transferencia de masas entre las fases y la cinética química.
Los recipientes de la reacción deberán ser resistentes a los ácidos, de fundición ó acero dotados de agitadores muy eficientes. Debido a la vigorosa turbulencia provocada, el área interfacial de la mezcla se mantiene tan alta como sea posible. Los reactores deben contener serpentinas internas para controlar la temperatura de una reacción altamente exotérmica.
Se puede producir nitrobenceno tanto en batch como en proceso contínuo. En los procesos típicos por lote, se carga el reactor con benceno y luego el ácido nitrante: 56-60% en peso de H2SO4, 27-32% de HNO3, y 8-17% de H20, se agrega lentamente debajo de la superficie del benceno. La temperatura de la mezcla se mantiene entre 50-55°C ajustando el caudal de alimentación de la mezcla ácida y del enfriamiento. La misma se puede elevar hasta 90°C hacia el final de la reacción para completarla. La mezcla reaccionada se pasa a un separador donde el ácido gastado sedimenta y se retira por el fondo, para ser reconcentrado-recuperado.
El nitrobenceno crudo sobrenadante se extrae por rebalse, es lavado en varas etapas con carbonato de sodio diluido y finalmente con agua. Según la pureza deseada del producto, se lo puede destilar.
Las necesidades de materiales para obtener 1000 kg. de nitrobenceno son:
| Benceno | C6H6 | 650kgs |
| Ácido Sulfúrico | H2SO4 | 720kgs |
| Ácido Nítrico | HNO3 | 520kgs |
| Agua | H2O | 110kgs |
| Carbonato de Sodio | CO3Na | 10kgs |
Usualmente un leve exceso de benceno se usa para asegurar que poco a ningún ácido nítrico quede en el ácido gastado. La duración de la reacción es generalmente de 2 a 4 hs y los rendimientos típicos son de 95-98% basados en el benceno cargado.
Un proceso de nitración contínua ofrece generalmente menores costo de capital, y un uso más eficiente de la mano de obra. Por eso, la mayoría de los productores de nitrobenceno emplean nitraciones contínuas. La secuencia básica de operaciones es la misma que para el batch, sin embargo, para una tasa de producción dada, el tamaño de los nitradores es mucho menor. Un nitrador contínuo de 0,114m3 posee la misma capacidad que un reactor batch de 5,68m3. A diferencia del proceso batch, el contínuo utiliza una menor concentración de ácido nítrico, y, debido al rápido y eficiente mezclado en los reactores pequeños, se observan altas tasas de producción.
Se alimentan benceno y el ácido nitrante :56-65% en peso de H2SO4, 20-26% de HNO3, y 15-18% de H20 dentro del reactor. Este puede ser un cilindro alargado con serpentinas internas de enfriamiento y sistema centrado de agitación. También se puede emplear un reactor tubular (tipo de casco y tubo con eficiente enfriamiento) que produzca un flujo turbulento. Generalmente la reacción se bombea a través del reactor en un lazo de recirculación, y se va retirando una porción de la mezcla y enviada al separador.
Usualmente se alimenta un leve exceso de benceno para asegurar que el ácido nítrico se consuma al máximo grado posible, y para minimizar la formación de dinitrobeneceno.
Se mantiene la temperatura del nitrador entre 50 a 100°C mediante el caudal de enfriamiento. La mezcla ya reaccionada fluye del nitrador al separador ó la centrífuga donde se separan las 2 fases. La fase acuosa ó ácido gastado sedimenta y se retira por el fondo a un concentrador de ácido sulfúrico, o bien se recicla al nitrador donde se mezcla con ácido nítrico y sulfúrico, inmediatamente antes de su ingreso al nitrador.
El nitrobenceno crudo fluye a través de varios lavadores separadores en serie donde el ácido residual se retira por lavado con una solución de carbonato de sodio seguida por un enjuague final con agua.
El producto luego se destila para quitar el agua y el benceno, y, si fuera exigido, se lo destila al vacío.
Los tiempos de reacción típicos son de 10-30 minutos, los rendimientos teóricos son de 96 - 99%. Generalmente se trata a las aguas de lavado con benceno mismo para extraerle el nitrobenceno, para el benceno que queda en las aguas de lavado se efectúa una operación de stripping previo a la planta de tratamiento de desagües.
Especificaciones del Nitrobenceno Dobledestilado
| Pureza | >99,8% |
| Color | claro amarillento |
| Punto de congelamiento | >5,13°C |
| Primera gota destilación | >207°C |
| Ultima gota destilación | 212°C |
| Humedad | <0,1% |
| Acidez (como HNO3) | <0,001% |
Características generales:
El nitrobenceno es una sustancia muy tóxica. La concentración máxima permitida es de 1ppm ó 5mg/m3. Es fácilmente absorbible por la piel y por inhalación de los vapores. El efecto primario es la conversión de la hemoglobina en metahemoglobina , lo cual inhibe el transporte de oxígeno en sangre, la cianosis se produce luego de 1 a 4 horas de latencia. La exposición continuada al nitrobenceno ataca al S.N.C. produciendo irritación de piel y ojos, fatiga, dolor de cabeza, pudiendo llegar al coma. Se deberán cambiar de ropas inmediatamente si resultaran manchadas; la piel salpicada debería ser lavada con agua caliente y jabón.
Está clasificado como de riesgo moderado para exposición al calor ó fuego.
El 97% en peso del nitrobenceno se convierte en anilina. De ésta anilina la mitad se emplea en la producción de MDI, un tercio en gomas químicas, 5% en tintas, 5% en la producción de hidroquinona. El nitrobenceno también se usa como solvente y en la refinación de aceites lubricantes.
El uso de nitrobenceno como disolvente de procesamiento en las reacciones químicas específicas es menor pero importante. La mayoría (95% o más) nitrobenceno producido se convierte en la anilina, que tiene cientos de productos secundarios. De ésta anilina la mitad se emplea en la producción de MDI, un tercio en gomas químicas, 5% en tintas, 5% en la producción de hidroquinona.
Otros usos industriales más bajos en volumen, pero sin embargo importantes, incluyen la reducción electrolítica de 4-aminofenol, nitración para dar 1,3-dinitrobenceno, la cloración para dar 3-cloronitrobenceno, sulfonación para dar ácido 3-nitrobencenosulfónico, y clorosulfonación para dar cloruro de 3- nitrobencenosulfonilo. Los tres últimos productos se consumen principalmente como sus productos de reducción, 3-cloroanilina, ácido metanílico y 3- aminobencenosulfonamida, respectivamente.